
Great running guys!
Congratulations to Scott Rosenberg on his PhD defense!
On December 11, Scott Rosenberg gave an impressive talk to finish out an impressive PhD thesis. He'll be returning to the Bay Area in January - we'll be sad to see him go. Scott's greatest hits (so far) can be seen here and here, and here and here (and there's more coming). Congratulations again Scott!
Congratulations to Alan West on a successful PhD defense!
Alan West successfully defended his PhD thesis on December 4, passing with flying colors! He spent most of his talk discussing his recently-published work on Hop1 and its dynamic behaviors in vitro, which you can read about in Nucleic Acids Research. Congratulations again, Alan!
Three thousand!

The main lab clone database just hit 3000 clones - great work guys!
Kevin Corbett profiled in Journal of Cell Science
Kevin sat down with Manuel Brewer at the Journal of Cell Science to discuss science, life, and starting a new lab. Read all about it!
Watching TRIP13 at work

When a cell gets ready to divide, it needs to make sure all of its chromosomes are lined up and ready to go, so none of them gets left behind. The Spindle Assembly Checkpoint monitors this process, and two key elements of the checkpoint pathway are the proteins MAD2 and TRIP13. In a new paper just published in The EMBO Journal, we reveal the molecular mechanism of a key step in this checkpoint pathway: its inactivation. We show that TRIP13 binds and pulls on MAD2's N-terminus to unfold the protein and disassemble the MAD2-containing mitotic checkpoint complex. Collaborating with Attila Toth in Dresden, we show that TRIP13 acts on the MAD2-related meiotic HORMADs using the same mechanism. Also contributing to the project were the labs of Don Cleveland here at Ludwig San Diego, and Franz Herzog in Munich. Great work everyone!
Another job for Monopolin - regulating the SUMO pathway!
Big congratulations to Namit Singh and Jason Liang for their recently-published work in Genes & Development: Recruitment of a SUMO isopeptidase to rDNA stabilizes silencing complexes by opposing SUMO targeted ubiquitin ligase activity. If you're interested but unable to access the paper online, please get in touch. This project was a close collaboration with Huilin Zhou's lab, our neighbor at the Ludwig Institute for Cancer Research. Just the first of many!
Lab Logo spotted in Western United States


Our lab logo's love of Nalgenes and travel continues with Gas Works Park in Seattle Washington and Lake Hemet's eagle preserve in California! It's just a matter of time until it travels to the eastern half of the US.
The Structural Biology Data Grid

A pretty good frame from our structure of C. elegans PCH-2
Just published - a new paper describing the Structural Biology Data Grid, an effort to archive and make available the primary data underlying crystal structures, among other types of "big data". This is a really valuable effort that we are supporting 100% - you can find the diffraction datasets supporting all of our structures on their web page here.
Lab logo spotted in the Sierra Nevadas

Our new and improved lab logo went on a trip to Kearsarge Pass in the Sierra Nevada Range for Independence Day! Stay tuned to see where it pops up next!
The multifaceted roles of the HORMA domain in cellular signaling
Just published: our new review in The Journal of Cell Biology describing the diverse signaling roles of HORMA domains throughout eukaryotic biology. The article is open-access, and can be found at JCB's web site.
SBGrid Member Tales
Just published: SBGrid Member Tale featuring Kevin Corbett! I could not be happier to be a member of SBGrid: extremely good support of all structure applications we could ever need, plus initiatives to bring big computing to structural biology in interesting ways. Enjoy!
Fun with animated GIFs
Just discovered GIFBrewery, which will convert movies into animated GIFs. This is pretty sweet for protein structure images on the web, as you can see below. Totally worth $5.

How TRIP13 inactivates the spindle Assembly checkpoint
Congratulations to Qiaozhen and the rest of the lab on the recent publication of our work on Pch2/TRIP13, a AAA+ ATPase that functions in both meiotic recombination control and in the spindle assembly checkpoint. The key result of this paper is that TRIP13, with the help of the adapter protein p31(comet), can actually convert the MAD2 protein from its signaling-active "closed" state to its inactive "open" state. We propose that this conformational conversion underlies TRIP13-mediated spindle assembly checkpoint inactivation. In the future, we will be working on how Pch2/TRIP13 might use a similar activity to regulate the conserved HORMAD proteins in meiosis (see our earlier work in Developmental Cell on these proteins), and we hope to also identify any additional substrates for TRIP13. You can read all about it at eLife:
TRIP13 is a protein-remodeling AAA+ ATPase that catalyzes MAD2 conformation switching
P.S. I must say that submitting to eLife was a pleasure. The reviews were among the most measured, helpful, and constructive I've ever experienced. Highly recommended, especially to fellow junior faculty!
Best Friends

Who says San Diego has no snow?
Art of the Gel Purification

HORMA domain proteins on the chromosome axis
I'm happy to share that the first major publication from the Corbett lab has been published in Developmental Cell! This paper describes the structures, interactions, and assembly of a family of proteins that bind chromosomes in meiosis, and control lots of different meiotic processes including formation of meiotic DNA breaks and their targeted repair into inter-homolog crossovers. This paper was a joint effort between our own Scott Rosenberg and Yumi Kim in the lab of Abby Dernburg at UC Berkeley and LBNL. It was truly a collaborative effort, combining the strengths of both of our labs. Thanks Yumi and Abby!
See the paper online at the link below. As always, if you can't access the publication, please contact me directly for a PDF reprint.
There are 8 structures associated with this paper, which will soon be made public on PDB.org. See our structures page for a summary.
Kim Y., Rosenberg S.C., Kugel C.L., Kostow N., Rog O., Dernburg A.F., Corbett K.D. (2014) The chromosome axis controls meiotic events through a hierarchical assembly of HORMA domain proteins. Developmental Cell (published online November 6, 2014). PRE-PUBLICATION JOURNAL LINK.
Monopolin directly cross-links sister kinetochores
We've thought for a long time (based on our crystal structures and other data from several labs) that the fungal monopolin complex directly cross-links sister kinetochores to ensure proper chromosome segregation in meiosis, but we were never able to directly prove it. Now, in collaboration with Chip Asbury and Adèle Marston's labs, we have directly demonstrated kinetochore cross-linking by monopolin in a single-particle setup. Pretty sweet!
Check out the paper at Science Express:
Sister kinetochores are mechanically fused during meiosis I in yeast
Krishna K. Sarangapani, Eris Duro, Yi Deng, Flavia de Lima Alves, Qiaozhen Ye, Kwaku N. Opoku, Steven Ceto, Juri Rappsilber, Kevin D. Corbett, Sue Biggins, Adèle L. Marston, Charles L. Asbury
Update: published in the 10 October 2014 issue of Science: PUBMED LINK
Now that's what I like to see when I walk into the lab...

Robust plasmid mutagenesis using isothermal assembly
Sometimes it seems like about half the time I spend in lab is time spent doing mutagenesis (yes, I am my lab's technician, and I like it that way). Quikchange (Now apparently "Quikchange Lighting") is the old standby, but now we have Q5 mutagenesis, and many other options I haven't tried. The drawback to all of these is that they rely on getting a polymerase to amplify an entire plasmid backbone, which is not a simple thing to do. I recently learned (from Chris Campbell in Arshad Desai's lab upstairs) about a way to use Gibson Assembly to make mutagensis, or by extension short insertions or deletions, a whole lot easier by breaking one long PCR into two shorter ones.
When I do mutagenesis, I typically start by using PrimerX to design a pair of complementary primers that should (and sometimes do!) work in a Quikchange-style PCR: 18-24 cycles of PCR, with a long extension time to allow the polymerase to synthesize the entire plasmid backbone. But more often than not, this doesn't work the first, second, or even third time I try it.

A primer pair designed for Quickchange Mutagenesis
What Chris realized was that almost every plasmid we use has a common feature: the antibiotic resistance gene. Chris designed a pair of complementary primers on the Ampicillin resistance gene in pET3a-type vectors (we also made a pair that prime off the Kanamycin resistance gene in pET28b-type vectors).
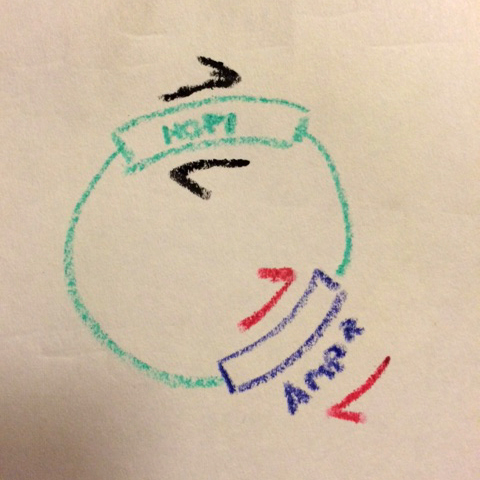
Two primer pars now, on opposite sides of the vector...
Then, for every mutation, he runs two reactions with different primer pairs:
(1) The forward primer for the mutation of interest + the reverse primer (#2 below) for the antibiotic resistance gene,
(2) The reverse primer for the mutation of interest + the forward primer (#1 below) for the antibiotic resistance gene.
The PCR products for (1) and (2) are shorter than the whole plasmid, and in my experience so far the reactions are MUCH more reliable (we use Phusion Polymerase almost exclusively). What you are left with is two linear pieces of DNA that overlap by 20-30 bases on each end - exactly what Gibson Assembly is good at sticking together. Gel purify the products from (1) and (2), toss a few nanograms of each in a Gibson Assembly reaction, and transform. Voila, easier mutagenesis.

Boom.
(BTW, I realize that this type of thing becomes obvious once you realize the power of combining Gibson Assembly with creative PCR and primer design - part of the point of this post is to get you thinking about ways to get your own cloning tasks done easier. Intron deletion? Chimeras? Multi-part assembly? Designed-from-scratch biofuel producing superbug? All easier than you think, thanks to Daniel Gibson and Craig Venter. Go San Diego!)
Our primer sequences for Amp and Kan genes, which work anywhere from 45° to 65° annealing temperatures (check the orientation of these on your vector before you use them - our Kan gene is backwards w/r/t our inserts, but the primers for both Amp and Kan are designed so that you use #2 with the mutagenic forward primer, and #1 with the mutagenic reverse primer. Your mileage may vary...):
Isotherm_Amp_primer1 AGAATTATGCAGTGCTGCC
Isotherm_Amp_primer2 GGCAGCACTGCATAATTCT
Isotherm_Kan_primer1 GACAATTACAAACAGGAATCGAATGC
Isotherm_Kan_primer2 GCATTCGATTCCTGTTTGTAATTGTC
Great references for Gibson Assembly:
Gibson, D.G. et.al (2009). Nature Methods. 343-345.
Gibson, D.G. et al. (2010). Nature Methods. 901-903.
Product page and usage guide: New England Biolabs 2X Gibson Assembly Master Mix
Update 10/6/14:
SGI-DNA, a spin-off of Synthetic Genomics here in La Jolla, is now selling a Gibson Assembly kit that they say is much better than "the leading competitor". I haven't tried it, but you can judge for yourself... at the very least, it is less expensive than NEB's kit.